Plotting inflation in association test statistic due to population structure
Last updated: 2020-12-22
Checks: 6 1
Knit directory: popstruct_scripts/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can run wflow_publish to commit the R Markdown file and build the HTML.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20201202) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 5bce003. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: code/.DS_Store
Ignored: data/.DS_Store
Ignored: data/burden_msprime/
Ignored: data/burden_msprime2/
Ignored: data/gwas/
Ignored: data/ukmap/
Ignored: output/plots/
Untracked files:
Untracked: analysis/biasvaccuracy_prsascertainment.Rmd
Untracked: analysis/plotting_prs_sib_effects.Rmd
Untracked: analysis/plottingprs_distribution_gridt.Rmd
Untracked: analysis/plt_burden_association.Rmd
Untracked: analysis/plt_gwas_results_t9.Rmd
Untracked: analysis/plt_ukb_unrelated_prs.Rmd
Untracked: analysis/prs_wt_finemapping.Rmd
Untracked: code/burden_msprime/
Untracked: code/fine_mapping/
Untracked: code/germline_ibd/
Untracked: code/gwas/
Untracked: code/imputation/
Untracked: code/optimize_migration_rate/
Untracked: code/pca_plots/
Untracked: code/prs/
Untracked: code/qqplots/
Untracked: code/revisions/
Untracked: code/shared_scripts/
Untracked: code/sib_analysis/
Untracked: code/simulating_genotypes/
Untracked: code/simulating_phenotypes/
Unstaged changes:
Modified: README.md
Modified: analysis/Simulating_heritable_phenotypes.Rmd
Deleted: analysis/Simulating_heritable_phenotypes.nb.html
Modified: analysis/_site.yml
Modified: analysis/index.Rmd
Modified: analysis/plt_PCA.Rmd
Deleted: analysis/plt_PCA.nb.html
Modified: analysis/plt_burden_clustering.Rmd
Deleted: analysis/plt_burden_clustering.nb.html
Modified: analysis/plt_lambda_v_frequency_ukb.Rmd
Deleted: analysis/plt_lambda_v_frequency_ukb.nb.html
Deleted: burden_msprime/.ipynb_checkpoints/Untitled-Copy1-checkpoint.ipynb
Deleted: burden_msprime/.ipynb_checkpoints/Untitled-checkpoint.ipynb
Deleted: burden_msprime/Notes_burden_msprime.txt
Deleted: burden_msprime/Untitled-Copy1.ipynb
Deleted: burden_msprime/Untitled.ipynb
Deleted: burden_msprime/burden_association_tests_norecomb.Rmd
Deleted: burden_msprime/burden_association_tests_norecomb.nb.html
Deleted: burden_msprime/burden_clustering.Rmd
Deleted: burden_msprime/burden_clustering.nb.html
Deleted: burden_msprime/burden_illustration.R
Deleted: burden_msprime/burden_test.haps.npz
Deleted: burden_msprime/generate_burden/burden_association.py
Deleted: burden_msprime/generate_burden/burden_gini.py
Deleted: burden_msprime/generate_burden/burden_gwas.txt
Deleted: burden_msprime/generate_burden/generate_burden_t100.py
Deleted: burden_msprime/generate_burden/generate_burden_t9.py
Deleted: burden_msprime/generate_burden/msprime_genic_burden_gini_nointrons_g_rho.py
Deleted: burden_msprime/generate_burden/msprime_genic_burden_t_r_x.py
Deleted: burden_msprime/generate_burden/wrapper_burden_association.sh
Deleted: burden_msprime/generate_burden/wrapper_burden_gini.sh
Deleted: burden_msprime/generate_burden/wrapper_generate_burden.sh
Deleted: burden_msprime/genos_gridt100_l1e7_ss750_m0.05_chr1_20.rmdup.train.cm.200k.eigenvec
Deleted: burden_msprime/genos_gridt100_l1e7_ss750_m0.05_chr1_20.rmdup.train.re.all.eigenvec
Deleted: burden_msprime/iid_train.txt
Deleted: burden_msprime/pheno_gridt100_noge_s9k.train.1.txt
Deleted: burden_msprime/plt_burden_association_t100.Rmd
Deleted: burden_msprime/plt_burden_association_t100.nb.html
Deleted: burden_msprime/plt_burden_association_t9.Rmd
Deleted: burden_msprime/plt_burden_association_t9.nb.html
Deleted: burden_msprime/plt_burden_clustering.Rmd
Deleted: burden_msprime/plt_burden_clustering.nb.html
Deleted: fine_mapping/comparing_susie_effects.R
Deleted: fine_mapping/comparing_susie_vs_ct.R
Deleted: fine_mapping/finemap.R
Deleted: fine_mapping/generate_genomic_coordinates_for_windows.R
Deleted: fine_mapping/generate_ldmat.sh
Deleted: fine_mapping/prs_wt_susie.Rmd
Deleted: fine_mapping/prs_wt_susie.nb.html
Deleted: fine_mapping/prs_wt_susie.sh
Deleted: fine_mapping/susie.R
Deleted: fine_mapping/wrapper_susie.sh
Deleted: germline_ibd/make_grm.R
Deleted: germline_ibd/proc_germline.R
Deleted: gwas/grid/notes_on_subsetting_snps_from_tau9.txt
Deleted: gwas/grid/tau-9/blmm.sh
Deleted: gwas/grid/tau-9/gcta_mlma_gridt9.sh
Deleted: gwas/grid/tau-9/gwas_wrapper_gridt-9_noge.sh
Deleted: gwas/grid/tau-9/gwas_wrapper_gridt9_ge.sh
Deleted: gwas/grid/tau-9/gwas_wrapper_gridt9_ge_geo.sh
Deleted: gwas/grid/tau-9/gwas_wrapper_gridt9_ge_re2.sh
Deleted: gwas/grid/tau-9/gwas_wrapper_gridt9_ge_repruned2.sh
Deleted: gwas/grid/tau-9/paste_cmre_pca.sh
Deleted: gwas/grid/tau-9/plot_prs_all.R
Deleted: gwas/grid/tau-9/processgwas4qq.R
Deleted: gwas/grid/tau-9/prs_wrapper.sh
Deleted: gwas/grid/tau-9/prs_wrapper2.sh
Deleted: gwas/grid/tau-9/prs_wrapper3.sh
Deleted: gwas/grid/tau-9/scripts/generate_genotypes/pca.sh
Deleted: gwas/grid/tau-9/scripts/generate_genotypes/vcf2plink.sh
Deleted: gwas/grid/tau-9/scripts/gwas/gwas.sh
Deleted: gwas/grid/tau-9/scripts/prs/cal_prs.sh
Deleted: gwas/grid/tau-9/scripts/prs/cal_prs2.sh
Deleted: gwas/grid/tau-9/scripts/prs/cal_prs3.sh
Deleted: gwas/grid/tau-9/scripts/prs/clump.R
Deleted: gwas/grid/tau-9/scripts/prs/clump2.R
Deleted: gwas/grid/tau-9/scripts/prs/clump3.R
Deleted: gwas/grid/tau-9/scripts/simphenotype/simgeffects.R
Deleted: gwas/grid/tau-9/scripts/simphenotype/simphenotype_ge.R
Deleted: gwas/grid/tau-9/scripts/simphenotype/simphenotype_ge_wrapper.sh
Deleted: gwas/grid/tau-9/scripts/simphenotype/simphenotype_noge.R
Deleted: gwas/grid/tau-9/simphenotype_noge.R
Deleted: gwas/grid/tau-9/split_beds.R
Deleted: gwas/grid/tau-9/wrapper_processqq_gridt9.sh
Deleted: gwas/grid/tau100/blmm.sh
Deleted: gwas/grid/tau100/blmm_nopc.sh
Deleted: gwas/grid/tau100/cat_gwas_sib.sh
Deleted: gwas/grid/tau100/cat_prs_sibs.sh
Deleted: gwas/grid/tau100/fastgwa.sh
Deleted: gwas/grid/tau100/gcta_mlma_gridt100_ge.sh
Deleted: gwas/grid/tau100/gctaloco_mlma_gridt100_ge.sh
Deleted: gwas/grid/tau100/gctaloco_mlma_gridt100_noge.sh
Deleted: gwas/grid/tau100/gwas_ge_incombined_sample.sh
Deleted: gwas/grid/tau100/gwas_wrapper_gridt100_ge.sh
Deleted: gwas/grid/tau100/gwas_wrapper_gridt100_ge_test.sh
Deleted: gwas/grid/tau100/lmmloco_wrapper_gridt100_ge.sh
Deleted: gwas/grid/tau100/lmmloco_wrapper_gridt100_noge.sh
Deleted: gwas/grid/tau100/locowrap_ge.sh
Deleted: gwas/grid/tau100/locowrap_noge.sh
Deleted: gwas/grid/tau100/paste_cmre_pca.sh
Deleted: gwas/grid/tau100/plot_prs_all.R
Deleted: gwas/grid/tau100/plot_prs_all_t100.R
Deleted: gwas/grid/tau100/prs_wrapper.sh
Deleted: gwas/grid/tau100/prs_wrapper_mlma.sh
Deleted: gwas/grid/tau100/prs_wrapper_sibs.sh
Deleted: gwas/grid/tau100/prs_wrapper_sibs_ascertained.sh
Deleted: gwas/grid/tau100/scripts/generate_genotypes/pca.sh
Deleted: gwas/grid/tau100/scripts/generate_genotypes/vcf2plink.sh
Deleted: gwas/grid/tau100/scripts/gwas/gwas.sh
Deleted: gwas/grid/tau100/scripts/prs/ascertain_effects.R
Deleted: gwas/grid/tau100/scripts/prs/cal_prs.sh
Deleted: gwas/grid/tau100/scripts/prs/cal_prs_mlma.sh
Deleted: gwas/grid/tau100/scripts/prs/cal_prs_sibs.sh
Deleted: gwas/grid/tau100/scripts/prs/cal_prs_sibs_ascertained.sh
Deleted: gwas/grid/tau100/scripts/prs/clump.R
Deleted: gwas/grid/tau100/scripts/prs/clump_mlma.R
Deleted: gwas/grid/tau100/scripts/prs/clump_pcs0.R
Deleted: gwas/grid/tau100/scripts/prs/clump_sibs.R
Deleted: gwas/grid/tau100/scripts/simphenotype/simgeffects.R
Deleted: gwas/grid/tau100/scripts/simphenotype/simphenotype_ge.R
Deleted: gwas/grid/tau100/scripts/simphenotype/simphenotype_ge_wrapper.sh
Deleted: gwas/grid/tau100/scripts/simphenotype/simphenotype_noge.R
Deleted: gwas/investigating_prs_ns_complexdem.Rmd
Deleted: gwas/investigating_prs_ns_complexdem.nb.html
Deleted: gwas/investigating_prs_ns_complexdem2.Rmd
Deleted: gwas/investigating_prs_ns_complexdem2.nb.html
Deleted: gwas/investigating_prs_ns_structure.Rmd
Deleted: gwas/investigating_prs_ns_structure.nb.html
Deleted: gwas/ukb/.ipynb_checkpoints/Untitled-checkpoint.ipynb
Deleted: gwas/ukb/Untitled.ipynb
Deleted: gwas/ukb/gwas_wrapper_ukb_ge.sh
Deleted: gwas/ukb/paste_cmre_pca.sh
Deleted: gwas/ukb/paste_cmre_pca_ukb.sh
Deleted: gwas/ukb/prs_wrapper.sh
Deleted: gwas/ukb/scripts/gwas/gwas.sh
Deleted: gwas/ukb/scripts/prs/cal_prs.sh
Deleted: gwas/ukb/scripts/prs/clump.R
Deleted: gwas/ukb/scripts/simphenotype/simgeffects.R
Deleted: gwas/ukb/scripts/simphenotype/simphenotype_ge.R
Deleted: gwas/ukb/scripts/simphenotype/simphenotype_ge_wrapper.sh
Deleted: gwas/ukb/scripts/simphenotype/simphenotype_noge.R
Deleted: gwas/ukb/scripts/simphenotype/simphenotype_noge_wrapper.sh
Deleted: imputation/extract_beagle_info.sh
Deleted: imputation/imputation_v_rarePCA.Rmd
Deleted: imputation/imputation_v_rarePCA.nb.html
Deleted: imputation/pca_on_imputed_genotypes.sh
Deleted: imputation/wrapper_beagle.sh
Deleted: imputation/wrapper_imputation.sh
Deleted: optimize_migration_rate/Fst_plots.R
Deleted: optimize_migration_rate/bplace_gwas.R
Deleted: optimize_migration_rate/complex_dem/bplacegwas_fst_grid.sh
Deleted: optimize_migration_rate/complex_dem/cal_fst.py
Deleted: optimize_migration_rate/complex_dem/complex_dem.py
Deleted: optimize_migration_rate/complex_dem/complex_dem_2.py
Deleted: optimize_migration_rate/complex_dem/complex_dem_bplace_wrapper.sh
Deleted: optimize_migration_rate/complex_dem/opt_lambda_complexdem.Rmd
Deleted: optimize_migration_rate/complex_dem/opt_lambda_complexdem.nb.html
Deleted: optimize_migration_rate/grid/tau-9/grid_bplace_wrapper.sh
Deleted: optimize_migration_rate/grid/tau100/grid_bplace_wrapper.sh
Deleted: pca_plots/Effect_of_using_cmre_together_pca.Rmd
Deleted: pca_plots/collinearity_bw_cmandrare_pcs.Rmd
Deleted: pca_plots/collinearity_bw_cmandrare_pcs.nb.html
Deleted: pca_plots/plt_complex_pca.R
Deleted: pca_plots/plt_pca.R
Deleted: prs/analyze_true_geneticeffects_out_o_sample.Rmd
Deleted: prs/biasvaccuracy_prsascertainment.Rmd
Deleted: prs/biasvaccuracy_prsascertainment.nb.html
Deleted: prs/clump_3.R
Deleted: prs/complex_dem/Plotting_esizes_and_prs.Rmd
Deleted: prs/complex_dem/Plotting_esizes_and_prs.nb.html
Deleted: prs/complex_dem/investigating_ns_strat.R
Deleted: prs/complex_dem/plotting_prs_from_sibeffects.Rmd
Deleted: prs/complex_dem/plotting_prs_from_sibeffects.nb.html
Deleted: prs/complex_dem/plottingprs_distribution_complex.Rmd
Deleted: prs/complex_dem/plottingprs_distribution_complex.nb.html
Deleted: prs/grid/plottingprs_distribution_gridt.Rmd
Deleted: prs/grid/plottingprs_distribution_gridt.nb.html
Deleted: prs/grid/tau100/Plotting_esizes_and_prs.Rmd
Deleted: prs/grid/tau100/Plotting_esizes_and_prs.nb.html
Deleted: prs/grid/tau100/plotting_prs_mlma.Rmd
Deleted: prs/grid/tau100/plotting_prs_mlma.nb.html
Deleted: prs/grid/tau100/plotting_prs_sib_effects.Rmd
Deleted: prs/grid/tau100/plotting_prs_sib_effects.nb.html
Deleted: prs/grid/tau100/plottingprs_distribution_gridt100.Rmd
Deleted: prs/grid/tau100/plottingprs_distribution_gridt100.nb.html
Deleted: prs/plot_expvobs_prs_4.R
Deleted: prs/plot_r2_rlat_supplement.Rmd
Deleted: prs/plot_r2_rlat_supplement.nb.html
Deleted: prs/prs_test_wrapper.sh
Deleted: prs/simulating_genetic_effects_prs
Deleted: prs/ukb/plt_ukb_unrelated_prs.Rmd
Deleted: prs/ukb/plt_ukb_unrelated_prs.nb.html
Deleted: prs/ukb/plt_ukb_unrelated_prs_uniform.Rmd
Deleted: prs/ukb/plt_ukb_unrelated_prs_uniform.nb.html
Deleted: qqplots/GWAS_qqdetails.txt
Deleted: qqplots/fixed_effects/plt_gwas_results_t100_all.Rmd
Deleted: qqplots/fixed_effects/plt_gwas_results_t9_07062020.Rmd
Deleted: qqplots/fixed_effects/plt_gwas_results_t9_07062020.nb.html
Deleted: qqplots/fixed_effects/plt_gwas_results_ti_all.Rmd
Deleted: qqplots/fixed_effects/plt_lambda_v_frequency.Rmd
Deleted: qqplots/fixed_effects/plt_lambda_v_frequency.nb.html
Deleted: qqplots/fixed_effects/plt_lambda_v_frequency_ukb.Rmd
Deleted: qqplots/fixed_effects/plt_lambda_v_frequency_ukb.nb.html
Deleted: qqplots/fixed_effects/scripts/plot_panels.R
Deleted: qqplots/fixed_effects/scripts/plot_panels_t100.R
Deleted: qqplots/fixed_effects/scripts/plot_panels_t9.R
Deleted: qqplots/fixed_effects/scripts/processgwas4qq.R
Deleted: qqplots/fixed_effects/scripts/wrapper_processqq_gridt100.sh
Deleted: qqplots/lmms/plt_gridt100_blmm.Rmd
Deleted: qqplots/lmms/plt_gridt100_blmm.nb.html
Deleted: qqplots/lmms/plt_gridt100_mlma.Rmd
Deleted: qqplots/lmms/plt_gridt100_mlma.nb.html
Deleted: qqplots/lmms/plt_gridt9_mlma.Rmd
Deleted: qqplots/lmms/plt_gridt9_mlma.nb.html
Deleted: qqplots/lmms/processgwas4qq_lmm.R
Deleted: qqplots/lmms/wrapper_processqq_gridt100.sh
Deleted: revisions/PCA_v_frequency_bracket_gridt100.sh
Deleted: revisions/PCA_v_frequency_bracket_ukb.sh
Deleted: revisions/PCA_v_number_of_cm_variants.sh
Deleted: revisions/ascertainment_schemes_prs_prediction.Rmd
Deleted: revisions/ascertainment_schemes_prs_prediction.nb.html
Deleted: revisions/calculate_prs_with_discoveryeffects.sh
Deleted: revisions/comparing_gvalues.R
Deleted: revisions/compute_genetic_values.sh
Deleted: revisions/compute_prs_a1_r2_p3.sh
Deleted: revisions/compute_prs_a1_r3s_p2.sh
Deleted: revisions/compute_prs_a3s_r1_p2.sh
Deleted: revisions/compute_prs_a3s_r2_p1.sh
Deleted: revisions/figuring_out_prediction_accuracy.Rmd
Deleted: revisions/figuring_out_prediction_accuracy.nb.html
Deleted: revisions/figuring_out_prediction_accuracy2.Rmd
Deleted: revisions/figuring_out_prediction_accuracy2.nb.html
Deleted: revisions/germline_ukb.sh
Deleted: revisions/rm_rare.sh
Deleted: shared_scripts/ascertain_effects.R
Deleted: shared_scripts/cal_prs.sh
Deleted: shared_scripts/cal_prs_mlma.sh
Deleted: shared_scripts/cal_prs_sibs.sh
Deleted: shared_scripts/cal_prs_sibs_ascertained.sh
Deleted: shared_scripts/clump.R
Deleted: shared_scripts/clump_mlma.R
Deleted: shared_scripts/clump_sibs.R
Deleted: shared_scripts/gen_map.R
Deleted: shared_scripts/get_se.R
Deleted: shared_scripts/gwas.sh
Deleted: shared_scripts/re_estimate_effects.R
Deleted: shared_scripts/simgeffects.R
Deleted: shared_scripts/simphenotype_ge.R
Deleted: shared_scripts/simphenotype_ge_wrapper.sh
Deleted: shared_scripts/simphenotype_noge.R
Deleted: sib_analysis/complex_dem/.ipynb_checkpoints/Sibling gwas - practice-checkpoint.ipynb
Deleted: sib_analysis/complex_dem/Sibling gwas - practice.ipynb
Deleted: sib_analysis/complex_dem/cat_sibs.sh
Deleted: sib_analysis/complex_dem/edit_fam.R
Deleted: sib_analysis/complex_dem/generate_gvalue_sib.py
Deleted: sib_analysis/complex_dem/generate_gvalue_sib_wrap.sh
Deleted: sib_analysis/complex_dem/generate_sib_phenotypes.sh
Deleted: sib_analysis/complex_dem/gwas_sib_complex_wrapper.sh
Deleted: sib_analysis/complex_dem/make_sib_haplotypes.py
Deleted: sib_analysis/complex_dem/mate4sibs.py
Deleted: sib_analysis/complex_dem/sib_gwas.py
Deleted: sib_analysis/complex_dem/simphenotype_sibs_ge.R
Deleted: sib_analysis/complex_dem/wrapper_generate_sib_haplotypes.sh
Deleted: sib_analysis/grid/tau100/generate_gvalue_sib.py
Deleted: sib_analysis/grid/tau100/generate_gvalue_sib_wrap.sh
Deleted: sib_analysis/grid/tau100/generate_sib_phenotypes.sh
Deleted: sib_analysis/grid/tau100/gwas_sib_grid_wrapper.sh
Deleted: sib_analysis/grid/tau100/make_sib_haplotypes.py
Deleted: sib_analysis/grid/tau100/mate4sibs.py
Deleted: sib_analysis/grid/tau100/sib_gwas.py
Deleted: sib_analysis/grid/tau100/simphenotype_sibs_ge.R
Deleted: sib_analysis/grid/tau100/wrap_gwas_reps.sh
Deleted: sib_analysis/grid/tau100/wrapper_generate_sib_haplotypes.sh
Deleted: simulating_genotypes/grid/generate_genos_grid.py
Deleted: simulating_genotypes/grid/simulating_and_processing_genotypes_t100.txt
Deleted: simulating_genotypes/grid/simulating_and_processing_genotypes_t9.txt
Deleted: simulating_genotypes/grid/tau-9/generate_genos_gridt9_wrapper.sh
Deleted: simulating_genotypes/grid/tau-9/generate_popfile_t9.R
Deleted: simulating_genotypes/grid/tau-9/pca_t9.sh
Deleted: simulating_genotypes/grid/tau-9/vcf2plink_t9.sh
Deleted: simulating_genotypes/grid/tau100/generate_genos_gridt100_wrapper.sh
Deleted: simulating_genotypes/grid/tau100/generate_popfile_t100.R
Deleted: simulating_genotypes/grid/tau100/pca_t100.sh
Deleted: simulating_genotypes/grid/tau100/pca_t100_test.sh
Deleted: simulating_genotypes/grid/tau100/vcf2plink_t100.sh
Deleted: simulating_genotypes/ukb/generate_genos_ukb.py
Deleted: simulating_genotypes/ukb/generate_pop_ukb.R
Deleted: simulating_genotypes/ukb/pca_ukb.sh
Deleted: simulating_genotypes/ukb/uk_nuts2_adj.txt
Deleted: simulating_genotypes/ukb/uk_nuts2_adj_ids.txt
Deleted: simulating_genotypes/ukb/ukb_gengeno_wrapper_1.sh
Deleted: simulating_genotypes/ukb/vcf2plink_ukb.sh
Deleted: simulating_phenotypes/Simulating_heritable_phenotypes.Rmd
Deleted: simulating_phenotypes/Simulating_heritable_phenotypes.nb.html
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
library(data.table)
library(dplyr)
library(ggplot2)
library(cowplot)
library(patchwork)
library(rprojroot)
F =is_rstudio_project$make_fix_file()Introduction
Here, I will demostrate how to generate the QQplots shown in Fig. 2 of the paper. These QQplots are generated from the GWAS of non-heritable phenotypes so any inflation in the test statistic, which shows up as a deviation from the diagonal is because of residual stratification and not because of polygenicity.
Implementation
The GWAS output gives us the \(\chi^2\) statistic of association and the p-value. To generate the QQplots, we need to generate their expected values and the 95% CI under the null distribution.
QQplots can be slighlty noisy so I carried out GWAS on 20 independent simulations of the phenotype and averaged the test statistic across each for plotting. To automate this process and avoid loading the large GWAS statistics files into memory all at once, I `processed’ each GWAS output by:
- sorting by the p-value,
- adding a rank to each variant. This will be used to generate the expected p-value and the 95% CI.
- indicating whether the variant is common or rare.
This allowed me to drop a lot of unncessary information from the GWAS output (chromosome no. SNP ID, position etc.). The code used to generate these files can be found under code/qqplots and the resulting file was then read and the expected statistics calculated as shown here.
Write function to read in the GWAS result and calculate expected statistics.
#function to summarize
fsummarize=function(phenotype="smooth",
correction="pcs0"){
#read the dataframe
dat = fread(F(paste("data/gwas/grid/genotypes/tau-9/ss500/train/gwas_results/fixed_effects/noge/genos_gridt9_",
phenotype,".",
correction,
".all.txt.gz",sep="")))
colnames(dat) = c("fcat","ID","P","ix")
dat=dat[,.(fcat,P,ix),]
#calculate the expected p-value from the rank
#observed chi-squared statistic from the observed p-values
dat[,c("exp.p","obs.chi"):=list(
ix/(max(ix)+1),
qchisq(P,df=1,lower.tail = FALSE)
), by=fcat]
#calculate expected chi-squared and genomic inflation
dat[,"exp.chi":=qchisq(exp.p,df=1,lower.tail = FALSE)]
dat[,"lambda":=obs.chi/exp.chi]
dat=dat[,chi.percentile:=1-exp.p]
#calculate 95% CI of the expected p-value
dat[,lower.ci:=qbeta(0.025,
shape1=ix,
shape2 = max(ix)-ix),by=fcat]
dat[,upper.ci:=qbeta(0.975,
shape1=ix,
shape2 = max(ix)-ix),by=fcat]
dat = dat[, lapply(.SD,mean),
by = .(fcat,ix)]
return(dat)
}
#write function to generate the QQplots.
fplot=function(dat,title="No correction"){
plt1<-ggplot(data=dat)+
geom_ribbon(aes(x=-log10(exp.p),
ymin=-log10(lower.ci),
ymax=-log10(upper.ci),
fill=fcat),
alpha=0.2,
show.legend = FALSE)+
geom_line(aes(-log10(exp.p),
-log10(P),
color=fcat),
size=0.7,
alpha=0.5,
show.legend = FALSE)+
geom_abline(intercept=0,slope=1,color="black")+
scale_color_manual(values=c("#ff7f00","#377eb8"))+
scale_fill_manual(values=c("#ff7f00","#377eb8"))+
theme_bw()+
theme(panel.grid=element_blank(),
axis.text=element_text(size=10),
plot.title = element_text(hjust=0.5),
plot.background = element_blank(),
plot.margin = unit(rep(0.5,4), "pt"))+
labs(color="Freq.",
title=title)+
xlim(c(0,max.log10P))+
ylim(c(0,max.log10P))
plt.inset=ggplot()+
geom_line(data=dat[chi.percentile>0.999,],
aes(chi.percentile,
lambda,
color=fcat),
show.legend = FALSE,
size=0.5)+
annotate(geom="text",
x=0.9993,
y=0.9*max.lambda,
label="lambda[p]",parse=TRUE)+
theme_bw()+
theme(panel.grid.major.x = element_blank(),
legend.position="none",
axis.title=element_blank(),
panel.grid=element_blank(),
plot.background = element_blank(),
axis.text.x = element_text(hjust=0,size=9),
axis.text.y = element_text(size=9))+
scale_x_log10(limits=c(0.999,1),
breaks=c(0.999,1),
labels=c("0.999","1"),
position="top")+
scale_y_continuous(limits=c(0.99, round(max.lambda,2)),
breaks=c(1, round(max.lambda,2)),
position="left")+
labs(x="p")+
scale_color_manual(values=c("#ff7f00","#377eb8"))
plt.wt.inset<-ggdraw(plt1) +
draw_plot(plt.inset, x=0.3, y=0.08, height=0.4,width=0.7)
return(plt.wt.inset)
}Read in the GWAS files for the `smooth’ phenotype for each method of correction, add the expected statistics, and calculate genomic inflation.
dat1=fsummarize("smooth","pcs0")
dat2=fsummarize("smooth","cm")
dat3=fsummarize("smooth","re")
median.lambda=lapply(list(dat1,dat2,dat3),
function(x){
return(
x[chi.percentile>0.49 &
chi.percentile<0.51,
median(lambda),by=fcat])
})
names(median.lambda)=c("pcs0","common","rare")
median.lambda=bind_rows(median.lambda,.id="correction")
colnames(median.lambda)[3]="lambda1"
median.lambda correction fcat lambda1
1: pcs0 common 1.158366
2: pcs0 rare 1.129383
3: common common 1.023470
4: common rare 1.019286
5: rare common 1.007668
6: rare rare 1.005272Generate the QQplots for the smooth phenotype.
max.lambda=max(sapply(list(dat1,dat2,dat3),
function(x){
max( x[ which(x$chi.percentile>0.999), "lambda"])
}))
max.log10P=max(sapply(list(dat1,dat2,dat3),
function(x){
max(-log10(x$P))
}))
plt1=fplot(dat1,"No correction")
plt2=fplot(dat2,"Common PCA")
plt3=fplot(dat3,"Rare PCA")
plt_combined.sm=plt1+theme(axis.title.x=element_blank())+
plt2+
plt3+theme(axis.title.x=element_blank())
plt_combined.sm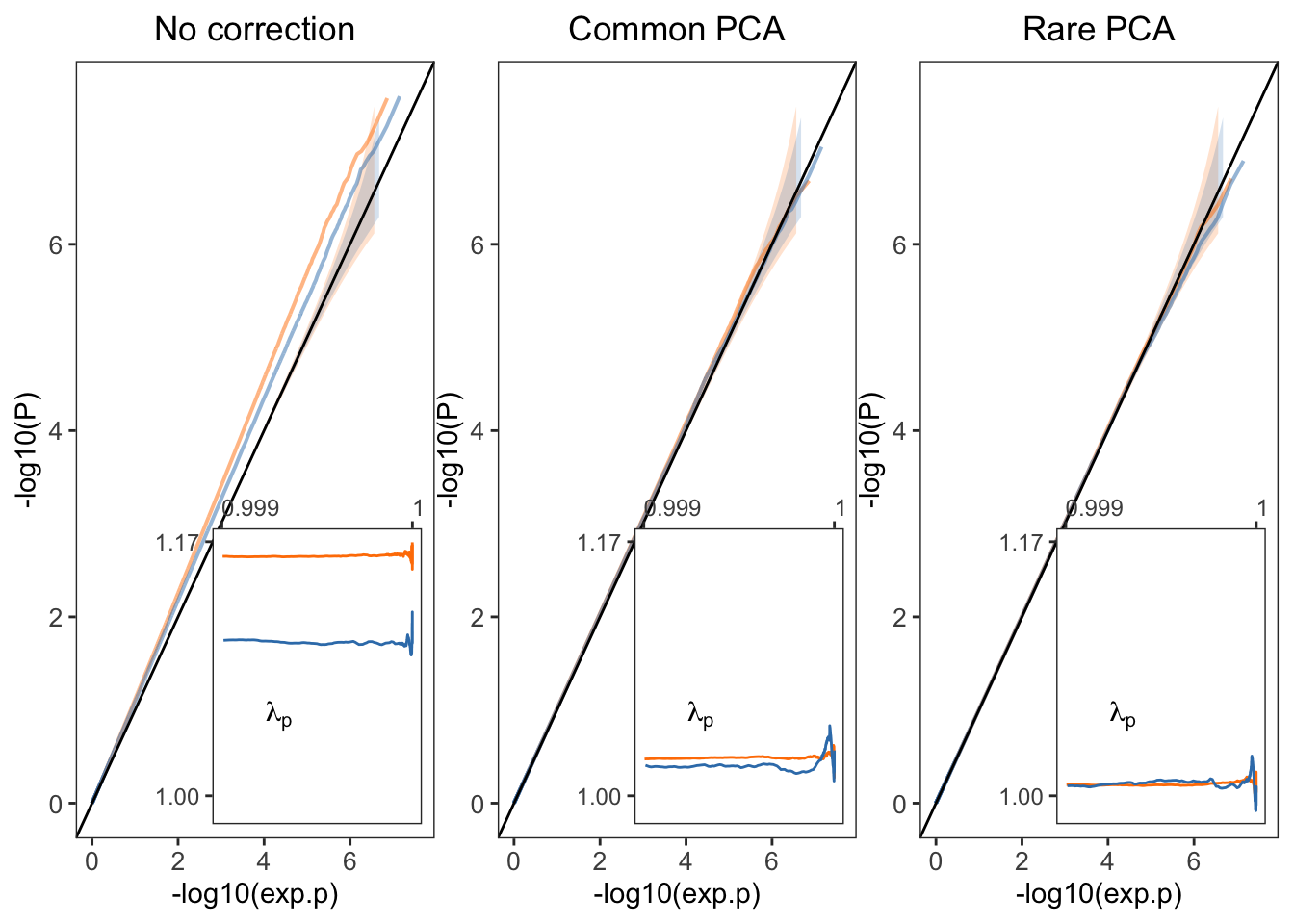
Plot for the sharp effect.
dat1=fsummarize("sharp","pcs0")
dat2=fsummarize("sharp","cm")
dat3=fsummarize("sharp","re")
median.lambda=lapply(list(dat1,dat2,dat3),
function(x){
return(
x[chi.percentile>0.49 &
chi.percentile<0.51,
median(lambda),by=fcat])
})
names(median.lambda)=c("pcs0","common","rare")
median.lambda=bind_rows(median.lambda,.id="correction")
colnames(median.lambda)[3]="lambda1"
median.lambda correction fcat lambda1
1: pcs0 common 1.0098105
2: pcs0 rare 0.9906909
3: common common 1.0071084
4: common rare 0.9918326
5: rare common 1.0055924
6: rare rare 0.9921018max.lambda=max(sapply(list(dat1,dat2,dat3),
function(x){
max( x[ which(x$chi.percentile>0.999), "lambda"])
}))
max.log10P=max(sapply(list(dat1,dat2,dat3),
function(x){
max(-log10(x$P))
}))
plt1=fplot(dat1,"No correction")
plt2=fplot(dat2,"Common PCA")
plt3=fplot(dat3,"Rare PCA")
plt_combined.shp=plt1+plt2+plt3
plt_combined.shp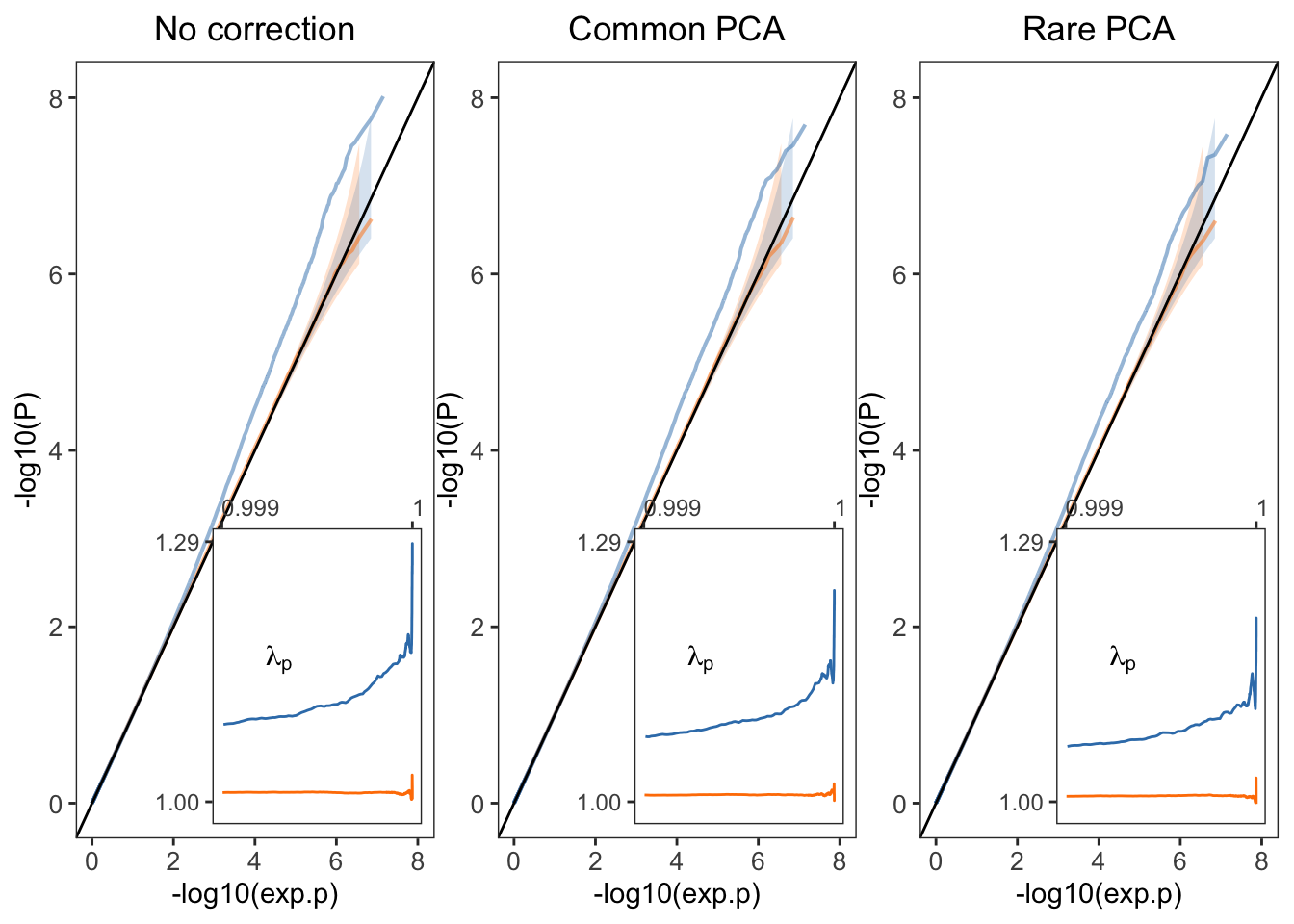
sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Catalina 10.15.7
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.0/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] rprojroot_1.3-2 patchwork_1.0.1 cowplot_1.1.0 ggplot2_3.3.2
[5] dplyr_1.0.2 data.table_1.13.2 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rcpp_1.0.5 pillar_1.4.6 compiler_4.0.3 later_1.1.0.1
[5] git2r_0.27.1 R.methodsS3_1.8.1 R.utils_2.10.1 tools_4.0.3
[9] digest_0.6.27 evaluate_0.14 lifecycle_0.2.0 tibble_3.0.4
[13] gtable_0.3.0 pkgconfig_2.0.3 rlang_0.4.8 rstudioapi_0.11
[17] yaml_2.2.1 xfun_0.19 withr_2.3.0 stringr_1.4.0
[21] knitr_1.30 generics_0.1.0 fs_1.5.0 vctrs_0.3.4
[25] grid_4.0.3 tidyselect_1.1.0 glue_1.4.2 R6_2.5.0
[29] rmarkdown_2.5 farver_2.0.3 purrr_0.3.4 magrittr_1.5
[33] backports_1.1.10 scales_1.1.1 promises_1.1.1 ellipsis_0.3.1
[37] htmltools_0.5.0 colorspace_1.4-1 httpuv_1.5.4 labeling_0.4.2
[41] stringi_1.5.3 munsell_0.5.0 crayon_1.3.4 R.oo_1.24.0